Terminal Sterilization of Sterile Filtered Products
The important shift towards terminal sterilization
Derek J. Prince, Ph.D.
Prince Sterilization Services, LLC
January 6, 2021
Terminal sterilization is the process of sterilizing products in their final container. With respect to pharmaceutical manufacturing, traditional liquid sterile drug products are filled into clean and sterile primary containers such as vials, bottles, syringes, cartridges, or bags. Post filling, the containers are closed off using primary and secondary packaging components such as pre-sterilized elastomeric stoppers, aluminum seals/closures, screw caps, or heat crimping methods. While strict controls related to current good manufacturing practices (cGMP) are employed to ensure the finished product is in fact sterile, no sterility assurance level (SAL) is obtained from this process. Therefore, the sterility of the drug product released from this type of manufacturing process is based entirely on the manufacturing controls in place at the time of filling. While USP <71> sterility release testing is often performed on a small percentage of units, the limitations associated with this test are well established [1].
Terminal sterilization is the only way to provide a SAL to a finished drug product. For this reason, it is well accepted that sterile drug products be manufactured using aseptic processing only when terminal sterilization is not an option [2]. With respect to a given sterilization modality, steam is the preferred method [3]. Heat lability of the packaging material should not in itself be used to disqualify a product from heat sterilization if it is otherwise heat stable [4]. If a decision is made to not terminally sterilize the drug product, drug manufacturers should be prepared to provide scientific explanation and justification for making such a decision [4]. Product degradation and container closure studies performed on product exposed to several different sterilization conditions (time/temperature) should be investigated prior to making a decision as to whether or not terminal sterilization is an option. A common misconception regarding terminal sterilization of drug products is that overkill type methods are required. This extreme level of sterilization is entirely unnecessary and not intended for product manufactured using aseptic manufacturing techniques where almost no bioburden exists [5]. As such, sterile drug manufacturers should not disqualify their product from terminal sterilization strictly based on the product not being able to withstand overkill type conditions. Instead, efforts should be made to identify the most product friendly sterilization conditions needed to establish a SAL that makes sense for the given product. The focus of the sterilization cycle must be on the ability of the process to kill bioburden organisms rather than unrealistically high populations of heat resistant spores (biological indicators; BIs) [5 & 6].
For making a decision related to the employment of terminal sterilization, referenced here are some further reading and a helpful decision tree (Figure 1) [7 & 8]. This decision tree emphasizes the importance of not disqualifying a product from terminal sterilization based on outdated overkill type sterilizing conditions. Instead, it provides several alternative terminal sterilization conditions to consider in the event that traditional sterilization temperature and time combinations are otherwise incompatible with the product in question. When reviewing this decision tree, it is important to remember that even a terminal sterilization cycle with an F0 of 2 (the equivalent of 2 minutes of sterilization exposure at 121°C) is significantly better than skipping a terminal sterilization step all together [9]. If providing a safe drug product for the patient is the ultimate intent of the numerous standards and regulations that are in place and adhered to by drug manufacturers, then it is imperative that terminal sterilization methods be utilized more frequently to achieve this goal.
In addition, less rigorous aseptic controls can be considered for drug manufacturers that terminally sterilize their final drug product [10 & 11]. This could translate to substantial savings as things like redundant filtrations and integrity tests/validations can be reduced and, clean room and personnel monitoring measures become less critical. Further, these reductions in aseptic controls could lead to even more significant reduced operating costs if certain personnel can be repositioned as they are no longer needed to perform intensive quality control and clean room monitoring tasks.
As a contract sterilizer, we feel passionate in our role in supporting the pharmaceutical industry and advocating for measures related to patient safety. We work closely with our customers to help establish a sterilization cycle that is compatible with their drug product, achieves a SAL, and is safe for the patient. In an effort to ensure that the terminal sterilization step is not cost prohibitive to the marketing of the drug product, we work hard to use the latest technology and process efficiencies to help offer extremely competitive pricing to all of our customers.
Prince Sterilization Services, LLC offers contract steam and dry heat sterilization and depyrogenation services to the pharmaceutical, medical device, and related health care industries. In addition, Prince also manufacturers the SteriKit®, a pharmaceutical quality kit containing ready to use vials, seals, and stoppers. Customers may supply their own components for processing or use components that we have in stock. Prince is FDA registered, ISO 13485:2016 certified, and cGMP compliant. Please reach out through our websites contact page or by contacting us using the link on this page.
www.princesterilization.com
info@princesterilization.com ; (888) 774-6230
Figure 1. A New Perspective Decision Tree
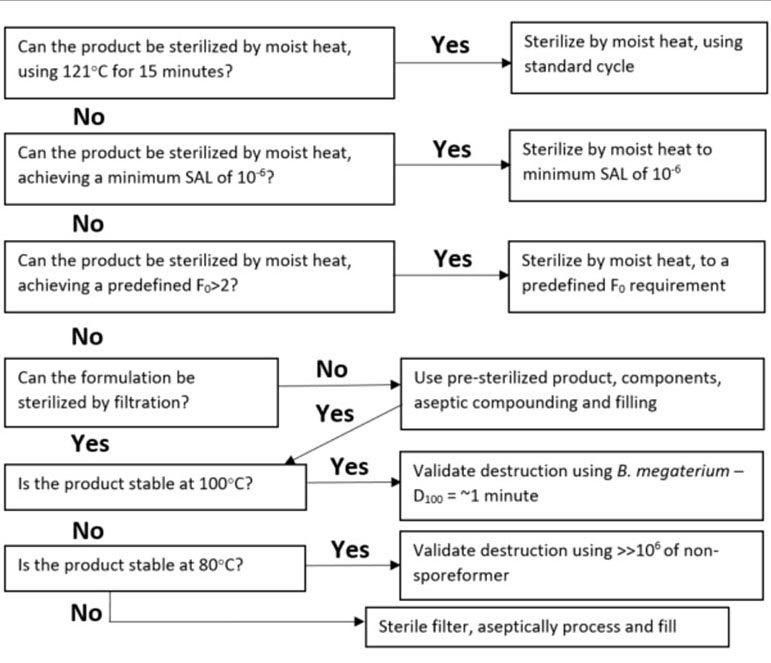
Adapted from Agalloco, J. [8]
References
- Sutton, Scott. The Sterility Tests. Microbiology Network, Inc. Rochester, NY. 2011.
- US Food and Drug Administration. Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. Rockville, MD: US Food and Drug Administration; 2004.
- EU GMP Annex 1: Manufacture of Sterile Medicinal Products (Draft), December 2020.
- European Agency for the Evaluation of Medicinal Products. Decision trees for the selection of sterilisation methods. London, United Kingdom: European Agency for the Evaluation of Medicinal Products; 2000
- United States Pharmacopeia. 43-NF 38, General Chapter <1211> Sterility Assurance. Bethesda, MD: United States Pharmacopeia; 2021.
- Prince DL, Prince DJ. Sterilization and Depyrogenation by Heat. In: Block SS, 6th ed. Disinfection, sterilization, and preservation. Philadelphia: Wolters Kluwer, 2021:589-608.
- Agalloco, J., “Redecorating the Tree”, published on-line at Linkedin.com, January 2020.
- Agalloco, J., Course notes, Sterilization: Principles & Validation, p. 159, 2018
- Sasaki T. Parametric release for moist heated pharmaceutical products in Japan. PDA Journal of GMP and Validation in Japan. 2002;4(1).
- US Food and Drug Administration. Compliance Program Guidance Manual Chapter 56 Drug Quality Assurance. 2015.
- United States Pharmacopeia. 43-NF 38, General Chapter <1116> Microbiological Control and Monitoring of Aseptic Processing Environments. Bethesda, MD: United States Pharmacopeia; 2021.